| HOME | HELP | CONTACT US | SUBSCRIPTIONS | CME | ARCHIVE | SEARCH | TABLE OF CONTENTS |
| ||||||||||||||||||||
Alex Tang | View/Change User Information | CiteTrack Personal Alerts | Subscription HELP | Sign Out
|
|
||||||||||||||||||||||||||||||
Alex Tang | View/Change User Information | CiteTrack Personal Alerts | Subscription HELP | Sign Out |
|||||||||||||||||||||||||||||||
(Pediatrics in Review. 2001;22:309-315.)
© 2001 American Academy of Pediatrics
 Professor of Pediatrics and Communicable Diseases;
Director, Division of Pediatric Endocrinology, University of Michigan
Medical School, Ann Arbor, MI.
Professor of Pediatrics and Communicable Diseases;
Director, Division of Pediatric Endocrinology, University of Michigan
Medical School, Ann Arbor, MI.
| Objectives |
|---|
|
|
|---|
| Introduction |
|---|
|
|
|---|
The diagnostic criteria for pubertal delay are based roughly on
statistical norms (ie, delay of more than 2 to 3 standard
deviations from the mean age of pubertal onset), but in fact they are
somewhat arbitrary. Puberty is considered to be clinically delayed if
sexual maturation has not become apparent by age 14 years in boys or
age 13 years in girls. This clinical diagnosis also is made in the
absence of menarche by age 16 years or in the absence of menarche
within 5 years of pubertal onset. Using these criteria, approximately
2.5% of healthy adolescents will be identified as having pubertal
delay. Most are boys. After evaluation, the majority of these
adolescents will be found to have no pathology; rather, onset of their
otherwise normal puberty simply is sufficiently late or slow relative
to that of their peers to have triggered concern and evaluation. When
puberty does begin, it is entirely normal. Some adolescents have a
variety of other causes to explain pubertal delay
(Table![]() ),
and with careful evaluation, most are diagnosed accurately.
),
and with careful evaluation, most are diagnosed accurately.
|
| Differential Diagnosis |
|---|
|
|
|---|
History may reveal similarly delayed puberty in the patient’s parents or siblings. Findings on physical examination are unremarkable except possibly for early signs of puberty unnoticed by the patient. Laboratory evaluation results are normal, although bone age is delayed and consistent with the extent of pubertal maturation.
The outcome of isolated constitutional delay of puberty is excellent; neither sexual maturity nor final adult height is affected by the timing of pubertal onset. However, when constitutional delay of puberty is superimposed on constitutional short stature, final height will be short. Resolving the relative effects of these factors in final height has been difficult in the studies performed to date.
Chronic Illness
Chronic illness may affect pubertal onset, tempo, and potential.
The pathophysiology of pubertal delay in chronic illness is variable,
frequently multifactorial, and in some cases, not well-established.
Chronic illness may affect underlying genetic potential, disturb
physiologic function, or limit adequate nutrition. Chronic use of
glucocorticoids, cancer chemotherapy, other medications, or radiation
therapy may have short- or long-term consequences for growth or sexual
maturation.
Adolescents who have chronic conditions, about which they already are acutely self-conscious, deserve particularly close monitoring of their pubertal course to allow early detection of pubertal difficulties. Often, only reassurance that puberty ultimately will proceed and eventual development will be normal is all that is required. However, when more pathologic pubertal derangements are detected, they should be treated early to maximize the potential for catch-up. Nutrition should be a priority, especially in those conditions where it frequently is compromised (eg, inflammatory bowel disease, cystic fibrosis), with supplementation used where appropriate. The risks and benefits of proposed medications and therapies, especially their effects on growth and maturation, always should be considered. Other explanations for pubertal delay should be investigated when the disease process does not explain maturational insufficiency or delay adequately. Finally, hormone replacement or augmentation with exogenous sex steroids is appropriate in some settings, but it is not a substitute for aggressive management of the underlying condition.
Hypopituitarism
PANHYPOPITUITARISM (CONGENITAL OR ACQUIRED).
Delayed puberty is not a common presentation of panhypopituitarism;
affected children typically present with short stature earlier in
childhood. Panhypopituitarism presenting in adolescence is usually due
to idiopathic hypothalamic failure. However, other unusual central
nervous system etiologies (eg, tumor, Langerhans cell histiocytosis)
should be ruled out.
ISOLATED GONADOTROPIN DEFICIENCIES (KALLMAN SYNDROME). The syndromes of isolated gonadotropin deficiency (IGD) are heterogeneous in clinical presentation. They occur more frequently in boys than in girls and often are difficult to distinguish from constitutional delay. IGD in association with hyposmia or anosmia is known as Kallman syndrome. The KAL-1 gene encodes a protein that allows fetal GnRH neurons to migrate from the olfactory placode through the cribiform plate and into the hypothalmus. Deletion of the KAL-1 gene is associated with sensorineural deafness, kidney malformations, and pes cavus. Boys who have GnRH deficiency often have a small phallus and testes, but findings on history and physical examination may be entirely normal except for sexual immaturity. Delayed bone age is the only consistent laboratory finding.
Other Endocrinopathies
HYPOTHYROIDISM.
Thyroid hormone is required for normal puberty. Its absence may delay
the onset or retard the progress of pubertal maturation by interfering
with gonadotropin secretion. Thyroid replacement therapy usually
normalizes gonadotropin secretion and allows puberty to proceed normally.
HYPERPROLACTINEMIA. Hyperprolactinemia may cause primary or secondary amenorrhea, but it is an otherwise rare cause of delayed puberty. Elevated prolactin levels interfere with gonadotropin production and may be due to a functioning pituitary adenoma (prolactinoma) or related to use of prescribed or illicit drugs (eg, phenothiazines, cocaine). Measurement of serum prolactin is a useful part of the evaluation for amenorrhea or delayed puberty, even in the absence of galactorrhea, and always should be obtained in the presence of galactorrhea. Prolactinomas may not be visible on imaging studies of the brain, making diagnosis more difficult.
Bilateral Gonadal Failure
Bilateral gonadal failure is uncommon and is characterized by
markedly elevated concentrations of serum gonadotropins. The
most common causes of gonadal failure are congenital: Turner syndrome
(gonadal dysgenesis) and Klinefelter syndrome. Other congenital causes
of gonadal failure and acquired bilateral gonadal failure are rare.
TURNER SYNDROME. Girls who have Turner syndrome have short stature, variable but incomplete puberty, primary amenorrhea, and characteristic congenital anomalies. Growth retardation is the most consistent characteristic and begins in utero. Rarely is a pubertal growth spurt seen. For some girls, the diagnosis will not be made until they present with pubertal insufficiency. Most girls who have Turner syndrome have primary ovarian failure that gives rise to markedly elevated levels of gonadotropins by adolescence, although variable sexual development still occurs. More than 50% of patients in one study had some breast development, and some pubic and axillary hair is typical for most patients. A minority of affected girls experience spontaneous menarche at an average age of 13.4 years, but most girls who have Turner syndrome require long-term estrogen replacement therapy.
KLINEFELTER SYNDROME. Klinefelter syndrome is relatively common. The genotype is typically 46,XXY, but genetic variability and mosaicism occur. Many affected boys are not identified until puberty or early adulthood. Often, some spontaneous pubertal development occurs, but testes become fibrotic and smaller as boys become older. Males who have Klinefelter syndrome present with small testicles and external genitalia and often have gynecomastia. Affected boys tend to be tall in childhood, and their tall stature sometimes delays diagnosis despite their pubertal immaturity. Borderline intellectual abilities or behavioral difficulties sometimes lead to diagnosis in childhood or may be appreciated in retrospect. In adolescence, young men who have Klinefelter syndrome present with small testicles and hypogonadism. Testosterone production is abnormally low, follicle-stimulating hormone values are high, and oligospermia or azospermia is seen.
ACQUIRED CAUSES OF GONADAL FAILURE. Iatrogenic gonadal failure may occur in the aftermath of chemotherapy, radiation therapy, or surgery. Acquired gonadal failure also may have traumatic, postinfectious, autoimmune, or metabolic causes. Mumps orchitis is the most common infectious cause of gonadal failure. Autoimmune oophritis, a rare cause of ovarian failure, often is associated with Addison disease and other autoimmune endocrinopathies. In galactosemia, the effects of galactose or its metabolites on the prenatal or neonatal ovary may cause delayed or deficient puberty in girls or may be a cause of menstrual dysfunction.
OTHER CONGENITAL SYNDROMES. Among patients who have complete androgen insensitivity, phenotypic females have the XY genotype and present with primary amenorrhea and sparse or absent pubic and axillary hair despite normal thelarche. Prader-Willi syndrome is associated with hypogonadism, short stature, and obesity in both males and females. Micropenis and bilateral cryptorchidism are characteristic of boys; hypoplasia of the labia majora and clitoris are seen in girls, who also frequently have delayed or absent menarche. Hypogonadotropic hypogonadism occurs in both the Laurence-Moon syndrome and the Bardet-Biedl syndrome. Boys who have Noonan syndrome have abnormal testes (cryptorchidism, atrophy, anorchia), and their sexual maturation is consistently delayed. Many have primary gonadal failure with no spontaneous puberty, and infertility is common.
In the "vanishing testes syndrome," 46,XY karyotype and masculine-appearing genitalia are associated with absent testes and failure of puberty. Presumably, testicular atrophy or destruction occurred some time after fetal differentiation of the external genitalia. Patients who have "resistant ovaries syndrome" have a 46,XX karyotype and typically present with sexual immaturity and primary amenorrhea. Further evaluation reveals small ovaries with primordial follicles despite elevated gonadotropin concentrations. The pathophysiology is believed to be due to abnormalities in gonadotropin receptors or antibodies to these receptors.
A long list of other congenital disorders and syndromes may be associated with pubertal delay or failure. These include Bloom syndrome, LEOPARD syndrome, ataxia telangectasia syndrome, and the cerebrohepatorenal syndrome. Enzyme defects in steroid synthesis (eg, cholesterol desmolase complex, 3-beta-hydroxysteroid dehydrogenase) also can lead to pubertal failure. Genetic males who have these disorders are born with ambiguous genitalia.
Hypogonadotropic Hypogonadism
Other conditions, including eating disorders, malnutrition, and
excessive exercise, may cause hypogonadotropic hypogonadism that
results in pubertal delay or insufficiency. Girls are affected more
often than boys and typically present with primary or secondary
amenorrhea. Girls who are competitive athletes have significantly later
pubarche and menarche than their peers, with the delays proportional to
the intensity of their training. Similarly, eating disorders can
disrupt normal pubertal progress profoundly. Weight gain usually
corrects these abnormalities, although women who have eating disorders
are at higher risk for menstrual irregularity independent of weight.
Congenital Anatomic Abnormalities
Congenital anomalies of the female reproductive tract usually
present with delayed onset of menses despite normal development of
secondary sexual characteristics. Congenital anomalies associated with
the apparent absence of menses include imperforate hymen, vaginal
atresia, or vaginal aplasia. Other girls present in the early teen
years with cyclic abdominal pain as a result of a normally responsive
endometrium and spillage of menstrual fluid into the pelvis
("concealed menarche").
| Evaluation of Pubertal Delay |
|---|
|
|
|---|
Determination of serum gondotropin levels will distinguish disorders of
congenital or acquired gonadal failure from other causes of delayed
puberty. By adolescence (bone age 10 to 12 y), gonadal
failure consistently produces markedly elevated levels of serum
gonadotropins. When these are present, findings from the history and
physical examination will often point to the specific diagnosis.
Chromosomal analysis is indicated to confirm clinical suspicion of
gonadal dysgenesis or Klinefelter syndrome. On the other hand, when
serum gonadotropins are normal or low, constitutional delay of puberty
is the most frequent diagnosis. However, further laboratory evaluation
may be required to exclude the possibility of occult chronic illness or
endocrinopathy. Reasonable screening studies include a complete blood
count, erythrocyte sedimentation rate, and measurement of serum
prolactin and serum thyrotropin-stimulating hormone. Even when results
of these studies are normal, IGD remains a possible diagnosis because
no studies reliably distinguish IGD from constitutional delay
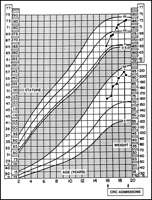
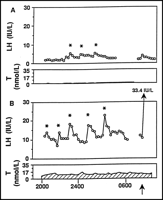
(Figs. 1![]() and 2
and 2![]() ).
Measurement of
first morning urinary testosterone or stimulation testing using a GnRH
agonist eventually may help to differentiate these conditions, but
neither study currently is in broad general use.
).
Measurement of
first morning urinary testosterone or stimulation testing using a GnRH
agonist eventually may help to differentiate these conditions, but
neither study currently is in broad general use.
|
|
| Management |
|---|
|
|
|---|
Testosterone injections (eg, testosterone enanthate 100 mg intramuscularly administered monthly for 6 mo) are used for boys whose pubertal development already has begun. For boys who have not yet begun puberty, oral oxandrolone can be used at a starting dose of 1.25 mg daily. Such treatment should be monitored by specialists familiar with the potential side effects, including hepatic peliosis and premature epiphyseal closure. All boys treated with sex steroids should be monitored at 3- to 6-month intervals to assess response to treatment and skeletal maturation. Treatment is discontinued when it is expected that endogenous hormone production is established.
Patients who have gonadotropin deficiency or hypogonadism require lifelong replacement with sex steroids and should be managed in consultation with a pediatric endocrinologist. Testosterone supplementation is begun in boys at approximately age 12 years. It may be administered by injection, transdermal patch, or topical gel. Doses initially are small and are increased based on careful follow-up of secondary sexual characteristics and growth to achieve a relatively normal puberty. Once puberty is complete, lifelong testosterone replacement is continued.
For girls who have hypogonadism, replacement hormone therapy is begun to coincide with puberty in peers. Estrogen replacement may be started with a transdermal estradiol patch or small daily doses of conjugated estrogens or ethinyl estradiol that are increased gradually to adult replacement levels. As with boys, careful monitoring of secondary sexual characteristics and growth is required. Cyclic hormonal replacement, typically with low-dose oral contraceptives, should be instituted after 1 to 2 years of estrogen replacement or once breakthrough bleeding has occurred.
| Psychosocial Consequences of Delayed Puberty |
|---|
|
|
|---|
Girls who have pubertal delay present with fewer psychosocial concerns. Indeed, some value their immature body habitus. For girls who have eating disorders, for example, early puberty consistently is associated with increasing dissatisfaction with weight or body image.
| Suggested Reading |
|---|
|
|
|---|
Blyth DA, Simmins RG, Zakin DF. Satisfaction with body image for early adolescent females: the impact of pubertal timing within different school environments. J Youth Adolesc. 1985;14:207
Brack CJ, Orr DP, Ingersoll G. Pubertal maturation and adolescent self-esteem. J Adolesc Health Care. 1988;9:280[CrossRef][Medline]
Brooks-Gunn J, Warren MP. The effects of delayed menarche in different contexts: dance and nondance students. J Youth Adolesc. 1985;14:285[CrossRef]
Ehrmann DA, Rosenfield RL, Cuttler L, Burstein S, Cara JF, Levitsky LL. A new test of combined pituitary-testicular function using the gonadotropin-releasing hormone agonist nafarelin in the differentiation of gonadotropin deficiency from delayed puberty: pilot studies. J Clin Endocrinol Metab. 1989;69:963[CrossRef][Medline]
Houchin LD, Rogol AD. Androgen replacement in children with constitutional delay of puberty: the case for aggressive therapy. Baillieres Clin Endocrinol Metab. 1998;12:427-440[CrossRef][Medline]
Kletter GB, Rolfes-Curl A, Goodpasture JC, et al. Gonadotropin-releasing hormone agonist analog (Nafarelin). A useful diagnostic agent for the distinction of constitutional growth delay from hypogonadotropic hypogonadism. J Pediatr Endocrinol Metab. 1996;9:9-19[Medline]
Lewis VG, Money J, Bobrow NA. Idiopathic pubertal delay beyond age fifteen: psychologic study of twelve boys. Adolescence. 1977;12:1[Medline]
Rosen DS. Pubertal growth and sexual maturation for adolescents with chronic illness or disability. Pediatrician. 1991;18:105[Medline]
Rosen D, Kletter GB, Kelch RP. Puberty: what to do when the clock doesn’t ring. J Pediatr Endocrinol. 1992;5:129
Click here for Delayed Puberty Author Disclosures Data Supplement
This article has been cited by other articles:
 |
D. S. Rosen Physiologic Growth and Development During Adolescence Pediatr. Rev., June 1, 2004; 25(6): 194 - 200. [Full Text] [PDF] |
||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HOME | HELP | CONTACT US | SUBSCRIPTIONS | CME | ARCHIVE | SEARCH | TABLE OF CONTENTS |