From Neonatal Network
Retinopathy of Prematurity: An Eye Toward Better Outcomes
Published: 06/29/2009
Abstract and Introduction
Abstract
Retinopathy of prematurity (ROP) results from the abnormal growth of blood vessels in the vascular bed supporting the developing retina. Estimated to cause up to 500 new cases of blindness in the U.S. each year, ROP affects primarily infants born at less than 1,500 g. Although its etiology is not well understood, ROP is thought to occur as a result of a complex interaction between oxygen and vascular growth factors. This article briefly reviews the history of ROP, discusses its pathophysiology, and addresses the risk factors and strategies for prevention.
Introduction
Premature delivery interrupts the orchestrated development of the fetus. Depending on the gestational age at the time of delivery, most organ systems in the newborn continue to develop. An exception is the vascular bed of the retina, the light-sensitive layer of cells of the eye. The fragile retinal blood vessels exposed to a hyperoxic extrauterine environment retreat and stop growing. Later, excessive vessel growth occurs in response to the hypoxia caused by the increased metabolic demands of the developing retina.[1]
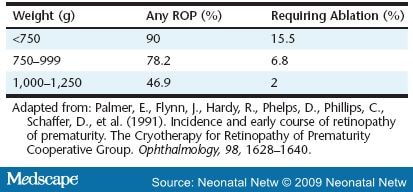
Retinopathy of prematurity (ROP) is the eye disease found in infants that results from this later, excessive, abnormal growth of new blood vessels (neovascular tissue) that nourish the retina. ROP is not initially a disease of the retina per se, but of the vascular bed that will one day support the retina. As the disease progresses, the abnormal capillaries invade the vitreous cavity, creating scar tissue that results in vision-threatening complications. This disorder usually develops in both eyes. It remains the second most common cause of childhood visual impairment and blindness in infants smaller than 1,500 g or very low birth weight (VLBW) in the U.S., with an estimated 500 infants blinded yearly and another 4,500 infants left with serious retinal scars ( Table 1 ).[1]
In order to evaluate research correlating oxygen delivery with the development of ROP, this author searched the Medline database from 2000 to 2006. The search strategy included the key terms infant, premature/blood; oxygen/adverse effects; low birth weight; infant, newborn; oxygen/administration and dosage; randomized controlled trials; retinopathy of prematurity/etiology; and retinopathy of prematurity/history. This summary of related research discusses the historical and clinical significance of ROP, as well as other risk factors of the disease. It correlates oxygen saturation values with the incidence of retinopathy. Current theories regarding the relationship between oxygen and growth factors responsible for the development of the vascular bed are explained. This article concludes with implications for nursing care.
History
In the 1940s, the incubator and the ability to deliver free-flowing oxygen to the infant within it were developed.[2] A once-rare malady began to surface with alarming frequency: the scarring and detachment of the retina caused by abnormal blood vessel growth. Over the next 12 years, 10,000 children succumbed to an epidemic of blindness, mostly in the U.S. In 1951, Dr. Kate Campbell correlated the toxic effects of oxygen with the development of the disease. She recommended avoiding the use of supplemental oxygen except in the treatment of cyanosis.[3] With the reduction of oxygen administration, the epidemic ceased. In the U.S. alone, the proportion of blindness fell from 50 percent in 1950 to 4 percent in 1965.[2,3] Unfortunately, this decline was accompanied by an increase in neonatal deaths and cerebral palsy.[3]
In the 1960s, blood oxygen monitoring became available, making it possible to monitor blood oxygen levels more precisely.[1] It was thought that ROP would become a problem of the past. However, today the disease is again on the rise as infants of even lower gestational age with less mature eye development are surviving.
Pathophysiology of ROP
Beginning at 16 weeks gestation, a fine meshwork of vessels that will support the growing retina begins developing from the optic disc and proceeding to the periphery inside the back of the fetal eye. The retina develops into a stratified neuron-rich sheet spreading to the outer edge of the eye, becoming fully vascularized by approximately 40 weeks postmenstrual age. Once vascularization is complete, the vessels are not susceptible to ROP. When an infant is born before term, the vascular growth may be interrupted, resulting in pathologic changes of the retina (Figure 1).
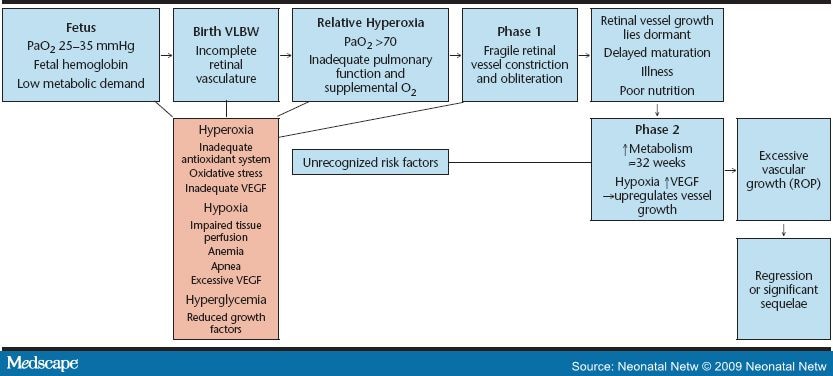
Figure 1. Progression of ROP.
ROP develops in two distinctly different phases. Phase 1 is short and almost immediate. Upon exposure to oxygen, the fragile new retinal vessels constrict to the point of obliteration. This may be an exaggeration of the protective measure in response to the higher oxygen now available to the tissues; however, it compromises retinal perfusion.[4] It also sets the stage for phase 2 of ROP. "The significance of oxygen levels lies in the nature of the circulation of the choroid layer, next to the retina, which is unique in that it fails to autoregulate in response to altered oxygen tension. Under hyperoxic conditions, the choroidal vessels cannot constrict, although the retinal vessels have this ability. As a result, excess oxygen moves from the choroidal to the retinal circulation, bathing the retina in oxygen and constricting the retinal vessels to the point of obliteration" (p. 697).[3]
Phase 2 begins as the infant matures. This phase of new vessel growth (neovascularization) occurs in response to the relative hypoxia caused by the increased metabolic demands of the developing retina.[1] In response to hypoxia, an overproduction of hormones and growth factors occurs; in particular, vascular endothelial growth factor (VEGF), but also growth hormones, including insulin-like growth factor 1 (IGF-1). These substances influence proteins of the extracellular matrix, such as fibronectin, vitronectin, and fibrinogen, to deposit adhesive fibrins and induce endothelial growth, differentiation, and migration. This new vessel growth can become poorly regulated and excessive, a condition termed prethreshold ROP. These vessels then become ectopic and begin growing three dimensionally into the vitreous space in the center of the eye, a condition called threshold ROP. Keshet explained that, rather than a gradual transition from vascularized to avascular retina, the retinal vessels terminate abruptly, marked by a demarcation line in the retina.[4] These abnormal vessels can begin to bleed and leak. This critical stage of ROP occurs most frequently at a postmenstrual age of 32--46 weeks, or 7--15 weeks postnatally.[5]
Linking Oxygen to ROP
Since the link was established between supplemental oxygen and ROP, there has been intensive research into the role of oxygen in the pathogenesis of this condition.[3] It is a matter of contention whether the amount of oxygen delivered or the length of oxygen exposure is the more important predisposing factor. A growing body of evidence implicates arterial oxygen fluctuations in influencing the disease.[6,7] Another theory is that the timing of exposure to oxygen is the critical factor. Hospitals vary widely in their incidence of ROP, even in like populations, and there is a remarkably wide variation in acceptable oxygen saturation levels in NICUs.[6,8] The effect of different target oxygen saturation ranges on retinal vascular growth is important, given the evidence that precise control of supplemental oxygen can reduce the incidence of ROP ( Table 2 ).

Vascular Endothelial Growth Factor
Mouse models have been used to study the pathogenesis of ROP. The mouse eye is incompletely vascularized at birth and resembles the eyes of premature infants. Exposure of newborn mice to hyperoxia causes obliteration of vessels and cessation of normal retinal growth, which mimics phase 1 of ROP in humans. When the mice are returned to room air, the nonperfused portions of the retina become hypoxic, which causes neovascularization similar to phase 2 of ROP.[9]
The recognition that hypoxia is the driving force of excessive retinal vessel growth prompted a search for a hypoxia-regulated chemical signal. One such chemical, was known to trigger blood vessel formation.[4,9,10] VEGF is detected in virtually all tissues, but is most abundant in the lung, kidney, and spleen. VEGF is fundamental for vessel growth and maintenance, and it regulates vessel permeability.[9,10] Smith discovered that VEGF is one of the most important growth factors involved in the pathogenesis of ROP.
She determined that, in phase 1 ROP, hyperoxia inhibits VEGF, thereby decreasing vessel growth, whereas in phase 2, retinal hypoxia stimulates an overexpression of causing excessive, rebound growth.[9]
Normally in the fetus, as the retina develops outward from the optic nerve, it encroaches into areas where there is no vasculature yet. The oxygen demand of vessel growth creates an area of physiologic (normal) hypoxia. In utero, the arterial oxygen pressure of the fetus is only 25 to 35 mmHg.[8,11] This physiologic hypoxia from low available oxygen stimulates the expression of anterior to the vessels, which grow toward the stimulus. When the newly formed vessels relieve the hypoxia in that area, VEGF is suppressed. The wave then moves forward as more retinal vessel growth begins.[9]
In infants born prematurely, the relative increase in oxygen extra utero interrupts this hypoxia wave and suppresses VEGF. Even if the infant requires only room air, his arterial oxygen pressure can rise to 60--100 mmHg, which is far higher than in utero values.[1] Normally, tissues respond to excess oxygen by pruning the microvasculature to match oxygen supply to their metabolic needs. However, in the eye, the excess oxygen is misinterpreted and results in a catastrophic overpruning of the newly formed vessels.[4] Furthermore, when hyperoxia suppresses VEGF to levels below what is needed for cell survival, it leads to apoptosis (cell death) in the vascular endothelial cells.[12] VEGF is further needed to repair these cells, but it is being suppressed by the hyperoxic state.
Insulin-like Growth Factor
Although VEGF is the most important contributor, blood vessel growth throughout the body is also dependent on other biochemical mediators. Insulin-like growth factor from the pituitary gland also promotes neovascularization by optimizing the activity of VEGF.[9,10] Studies show that a lack of IGF-1 prevents normal retinal vascular growth despite the presence of VEGF.[9,13]
IGF-1 is supplied by the placenta and is found in the amniotic fluid, which the developing fetus drinks.[9] After delivery, this supply is cut off. IGF-1 is also found in mother's milk. However, initially, most premature infants are not able to tolerate feedings. IGF-1 is a peptide and may be reduced further by poor nutrition, acidosis, low thyroxin production, and sepsis.[13] The loss of IGF-1 is associated with loss of vascular growth in phase 1 and proliferation of ROP in phase 2.[12] A persistently low serum concentration of IGF-1 in premature infants is considered to be a risk factor for ROP and may become a screening tool for ROP in the future.[13]
Providing IGF-1 after birth may allow faster maturation of the liver, the gastrointestinal tract, and other organs that may then produce the necessary factors for growth and development.[12] Increased protein intake increases IGF-1 levels in premature infants, so early breast milk feedings may be particularly beneficial because breast milk contains available IGF-1.[13] It also contains many antioxidant constituents, including inositol, vitamin E, and vitamin A.[14]
Metabolic Rate and Oxygen Consumption
The increased metabolic demands of extrauterine life require greater oxygen delivery. The neonate consumes twice as much oxygen as the adult in relation to weight. The metabolic demand and oxygen consumption in the premature neonate increase steeply with physical activity, a nonneutral thermal environment, procedures, and the work of breathing. All infants, and especially preterm infants, must be given adequate oxygen to meet their metabolic needs. Healthy term infants maintain a partial pressure of arterial oxygen (PaO2) of approximately 70 mmHg and 95 percent saturation within the first few hours of birth. This level exceeds the fetal demands for oxygen, yielding a relative hyperoxia in preterm neonates.[11] This can be damaging to the developing vascular bed of the retina and alveoli. To the immature infant, breathing even room air represents a hyperoxic exposure.[15] It is impossible to determine the physiologic PaO2 of an extremely preterm infant, but targeting ranges similar to well term infants may be inappropriate: "Although healthy preterm infants maintain an O2 saturation of 95 percent most of the time, artificial attempts to keep arterial oxygenation at 'physiological' level may do more harm than good in the first few weeks of life"(p. F110).[6]
Fetal Hemoglobin
Abundant oxygenation for growth of the fetus is made possible, in part, by fetal hemoglobin (HbF). The hemoglobin molecule in fetal blood is structurally different from its counterpart in the adult and carries a larger quantity of oxygen. The highest PaO2 in the fetus is in the umbilical vein blood leaving the placenta, usually about 30 mmHg. At that oxygen tension, the saturation of fetal blood is 6--8 percent higher than in the adult. During the first six months of life, HbF decreases from about 75 percent to about 2 percent of the total circulating hemoglobin. This decline in HbF is greater in the preterm infant and reaches its minimum by 3 to 12 weeks after birth. In other words, until the infant becomes anemic or receives adult blood, his HbF assures delivery of oxygen at oxygen saturation values 6 to 8 percent lower than the adult. This left shift of the oxygen dissociation curve affords some protection from hypoxemia and low cardiac output.[11] Consequently, York and colleages, and Saugstad recommended keeping oxygen saturations at the lower end of the safe range (85 percent) in the VLBW infant directly after delivery and possibly for the first few weeks of life.[7,10] In clinical practice, most clinicians follow the values of the oxygen saturation monitor and do not analyze fetal hemoglobin concentration and its potential involvement in shifts of the dissociation curve.[16]
Oxidative Stress
A growing body of research shows a correlation between oxidative stress and ROP, broncopulmonary dysplasia (BPD), necrotizing enterocolitis, and periventricular leukomalacia.[10,15,17,18] A by-product of normal cell metabolism is the formation of oxygen free radicals, highly reactive oxygen atoms that can attack and degrade chemical bonds in the cell membrane or nucleic acid.[19] Oxygen free radicals are produced in excessive amounts under hyperoxic conditions. They are extremely damaging to vascular endothelium and cells of the lung, retina, and brain. Free radicals are rendered inert by antioxidant enzymes that protect cells by scavenging and detoxifying the highly reactive oxygen metabolites.
The development of the biochemical antioxidant system occurs late in gestation.[10] The lower antioxidant defenses of premature infants make them more vulnerable to increases in oxygen free radical injury to the developing retinal vessels.
Antioxidents: Vitamins A and E
Low plasma vitamin A concentrations have been associated with the development of ROP. Vitamin A is necessary for orderly growth and differentiation of tissues. Approximately 90 percent of the body's vitamin A is stored in the liver, but the eye and the lung are other storage sites. In the retina, oxidation of vitamin A produces retinaldehyde, an essential component of the visual pigment necessary for visual response to light. In addition, the antioxidant properties of vitamin A may reduce damage to the developing retina from free oxygen radicals. Oral supplementation of 4,000 IU/kg/day is recommended for VLBW infants from establishment of enteral feedings until discharge.[20]
Vitamin E, another antioxident, has been studied in relationship to iron supplementation in erythropoesis and in the reduction of ROP. Although vitamin E supplementation may also be useful in decreasing the severity of ROP, its use has largely been abandoned because prolonged elevations in serum vitamin E levels were associated with an increased incidence of sepsis and necrotizing enterocolitis in VLBW infants.[21]
Risk Factors
Among the many proposed causes of ROP, "only low birth weight, low gestational age, and supplemental oxygen therapy following delivery have been consistently associated with the disease" (p. 134).[22] The earlier that delivery occurs, the more likely that ROP will result. ROP is rare in infants older than 32 weeks gestation and heavier than 1,500 g. However, 47 percent of infants with a birth weight between 1,000 and 1,250 g have some degree of ROP, and 90 percent of infants less than 750 g develop ROP.[23] A similar pattern is associated with early gestational age, with ROP occurring in 84 percent of infants born at less than 28 weeks.7 Factors associated with oxygen include frequent apnea, sepsis, long-term ventilator use, and anemia (Figure 2). Frequent apnea is cited as the most significant independent risk factor for ROP by Kim and associates, who state, "The fluctuation of arterial oxygen tension may induce local production of vasogenic factors which may subsequently lead to neovascularization and ROP. This process may be even more significant in babies who have preexisting, prethreshold ROP" (p. 133). They noted that patients who progressed to severe ROP had more frequent apnea than those whose retinopathy regressed.[24] Severe anemia may also play a role in increasing apnea episodes.[25] This creates greater oxygen starvation in an already oxygen-starved retina during phase 2 ROP.
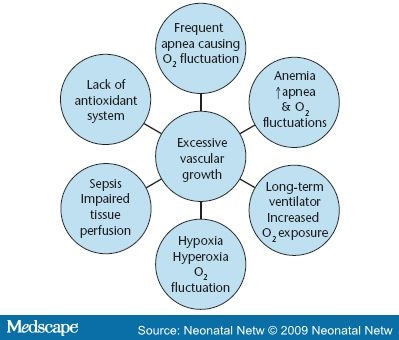
Figure 2. Oxygen assiciated risk factors in the development of ROP in the low birth weight/low gestational age infant.
Bacterial and yeast sepsis are frequently cited risk factors.[24,26] Sepsis is often accompanied by the systemic inflammatory response, including hypotension and inflammation of the vascular bed throughout the entire body, including the retina. The endothelium, or lining of the vascular bed, houses a dynamic matrix of neutrophils, cytokines, mediators, and hormones that respond to the invasion of bacteria and yeast. When these substances are released, capillary walls become dilated and permeable, allowing fluids and debris to leave the capillary bed, saturating the surrounding tissues, and causing inflammation, edema, and scar tissue. This process may impair tissue perfusion and release angiogenic factors secondary to hypoxic stress. Neutrophils responding to the damage adhere to the endothelium and undergo a sudden increase in oxidative metabolism, releasing oxygen free radicals. In addition, neutrophils are known to produce more than 50 different toxins that cause capillary damage.[19] Because the integrity of the capillary bed of the retina can be directly visualized with an opthalmoscope, this examination provides a glimpse into the general health of the baby.
Erti and coworkers claimed there is a relationship between hyperglycemia and ROP, noting that hyperglycemia plays an important role in the development of proliferative retinopathy in the diabetic adult. When they analyzed VLBW infants retrospectively, they found gestational age and hyperglycemia to be significant contributors to the disease. These observations alert clinicians to consider hyperglycemia (already known to result in osmotic diuresis, dehydration, weight loss, increased incidence of intraventricular hemorrhage, and increased mortality rate) to also be a potential risk factor for ROP development.[27]
The LIGHT-ROP clinical trial in 1999 tested the hypothesis that reducing the ambient light reaching the eyes of preterm infants may be effective in preventing ROP. The reasoning centers on the role of light in the production of free radicals. Supplemental oxygen also produces free radicals, and the two mechanisms may be additive. In a masked control study, 361 VLBW infants were divided into those wearing light-absorbing eye covers and those uncovered. The researchers found no difference in the amount or severity of ROP in either group.[28]
The smallest and sickest infants, with the most underdeveloped and fragile retinal vasculature, often require extended ventilator and oxygen therapy. The PaO2 can fluctuate rapidly in VLBW infants, leading to alternating episodes of severe and extended hyperoxemia and hypoxemia. These shifts in oxygenation are thought to trigger spikes in VEGF, resulting in abnormal retinal vessel growth.[18] Often oxygen fluctuation is related to the infant's underlying disease, but it may also be the result of routine medical intervention.[7] Armed with the knowledge of how oxygen affects VEGF, Chow and colleagues postulated that it is not only hyperoxia that encourages the development of ROP, but the repeated episodes of hypoxia-hyperoxia often seen in hard-to-manage, chronically ill infants. Hypothesizing that some of the variations in ROP rates between hospitals are related to the minute-to-minute handling of oxygen administration, they set up a strict oxygen management policy designed to avoid hyperoxia (defined as oxygen saturation values above 93 percent) as well as repeated episodes of hypoxia-hyperoxia in infants born less than 1,500 g. Over the next five years, their incidence of ROP decreased consistently from 12.5 percent to 2.5 percent. The Vermont Oxford Network currently recommends the practice of avoiding oxygen saturations above 93 percent and oxygen fluctuations.[8] The reduction in ROP may have been due to tighter oxygen control decreasing the fluctuations in saturations or due to the lower pulse oximeter saturation values (SpO2).[1]
Oxygen-saturation Targets
The use of supplemental oxygen has a long history in neonatal medicine, resulting in both significant health care benefits and harms. The dilemma is that although oxygen is essential for normal metabolism, VLBW infants are particularly sensitive to its toxic effects. Keeping in mind that phase 1 and phase 2 have opposite trends in retinal vascularization, the operative question is: What is the most appropriate range to target blood oxygen levels in the VLBW infant so as to minimize both phases? Potential benefits of higher oxygen levels in phase 2 include less apnea, better feeding, improved long-term growth and development, and more stable sleep patterns.[17] However, high oxygen levels may have severe deleterious effects on the retinal vascular system, especially during phase 1.
Oxygen Saturation Targets During Phase 1 ROP
A retrospective study by Tin and coauthors examined the case notes of 295 VLBW infants to determine whether different policies with regard to the control of SpO2 had any impact on developing ROP. The study found that those infants given enough supplemental oxygen to achieve SpO2 of 88--98 percent for at least the first 8 weeks of life developed threshold ROP four times as often as babies given only enough oxygen to maintain an SpO2 of 70--90 percent. No difference was found in the proportion who survived infancy or who later developed cerebral palsy.[6] This article suggests that using lower settings for alarms (SpO2 70--90 percent) in infants less than 28 weeks gestation for the first 8 weeks may have significant benefits for infants in terms of less respiratory support, less weight loss, and less ROP.
There has been concern raised that Tin and associates' suggestions could increase the risk of life-threatening events and the risk of pulmonary hypertension in infants with chronic lung disease.[29] Although Tin and associates found the cerebral palsy rate at 18 months of age unaffected, early prolonged hypoxemia has been associated with subsequent cognitive defects.[30] Tin and colleagues explained that no infant received hospitalized care with SpO2 levels of 70 percent, and they observed that nurses and respiratory therapists tend to keep oxygen saturations toward the upper half of the ordered parameters. The wider oxygen saturation parameters also minimized the tendency to frequently adjust oxygen delivery to meet fluctuating saturation values, thus minimizing reflexive overshooting of the target oxygen range. Although Tin and associates's study illustrated that lower alarm settings may benefit the retina, it is not currently recommended to accept saturation limits less than 85 percent due to concerns regarding oxygenation of other organs and tissues.[29-31]
York and coworkers carried out a retrospective cohort study of 257 VLBW infants examining the influence of arterial oxygen fluctuation on the development of ROP. Their work on human infants was supported by experiments in newborn rats that clearly demonstrated that PaO2 fluctuations were more damaging than constant oxygen exposure in the development of abnormal retinal vessel growth. York's group found that the variability of oxygen levels, especially in the first two weeks of life, was a significant predictor of severe ROP. Early gestational age and very low birth weight were important independent contributors to the development of ROP. The more immature the retinal vessels, the more vulnerable they are to the environment outside of the womb. The oxygen in arterial blood can fluctuate rapidly in sick premature infants, making them more difficult to manage without overshooting the target range of PaO2. York's group argued that better controlling infant PaO2 fluctuations may lead to a decreased incidence of ROP.[7]
Oxygen Saturation Targets During Phase 2 ROP
The neovascularization process of phase 2 probably starts at about 32 to 34 weeks postmenstrual age, regardless of gestation at birth.[9,17,23] The critical stage of ROP occurs when abnormal vessels begin to extend into the vitreous space around 36 weeks postmenstrual age.[7,23] Phase 2 excessive vessel growth is a consequence of hypoxia resulting from the vascular obliteration of phase 1. Few NICUs have specific guidelines for monitoring oxygen in relation to gestational and chronological age.[1] Two well-known trials, the STOP-ROP trial and the BOOST trial, have investigated the effect of using increased supplemental oxygen to reduce phase 2 ROP and to enhance infant somatic growth, respectively.[17,32]
STOP-ROP Trial. The STOP-ROP multicenter study group randomized 649 infants with prethreshold (moderately severe) ROP to receive supplemental oxygen to maintain SpO2 of 89--94 percent (control) or 96--99 percent (treatment). More infants in the control group (46 percent) progressed to threshold disease than infants in the treatment group (32 percent), a statistically significant difference. This suggests that liberalizing SpO2 targets once an infant reaches prethreshold ROP may improve retinal outcomes.[32] However, the benefit was lower than expected. Perhaps once an infant has reached prethreshold disease, it is too late for an increase in oxygen to relieve the hypoxia.
The study concluded, "The STOP-ROP data clearly demonstrate that oxygen at saturations of 96--99 percent does not increase the severity of ROP in eyes of infants with prethreshold ROP" (p. 307). However, it also showed a modest exacerbation of chronic lung disease and a lack of improvement in long-term growth and development at three months of age.[32] The poorer pulmonary outcome of the treatment group serves to remind us of the toxic effect of oxygen on developing tissue.
BOOST Trial. Askie and colleagues have studied the effects on growth and neurodevelopmental outcomes of maintaining SpO2 above 90 percent in preterm infants. The Benefits of Oxygen Saturation Targeting (BOOST) trial studied 358 infants of less than 30 weeks gestation who remained on oxygen at 32 weeks. Presuming that infants have a higher metabolic demand by 32 weeks, the researchers asked whether increased oxygen would affect somatic growth and mental development at 12 months corrected age. Secondary endpoints included the progression of ROP and the need for respiratory support. The control group received supplemental oxygen to achieve 91--94 percent SpO2. The treatment group received supplemental oxygen to achieve 95--98 percent SpO2.[17]
No significant differences in weight, length, or head circumference were found between groups at 12 months of age. The frequency of major developmental abnormalities did not differ. The high-saturation group had a higher rate of oxygen dependence at 36 weeks postconceptional age. However, the risk of developing severe ROP was less in the higher group (12 percent high vs 16 percent standard), as was the need for retinal surgery (6 percent high vs 11 percent standard). Although these differences were not statistically significant, the difference in the need for surgery approached significance in the subgroup of infants born prior to 28 weeks gestation (8 percent high vs 16 percent standard, p=.06).[17]
It is imperative to note that neither study accepted SpO2 of 100 percent. Once this value is reached, it is impossible to determine the arterial oxygen value or PaO2. When an infant's vascular bed is still growing (phase 2), care must be taken to prevent SpO2 of 100 percent. Hyperoxia remains the trigger for vasoconstriction and the consequent poorly regulated and excessive vessel growth. Oxygen saturation monitoring and careful regulation of oxygen delivery remain paramount until full vascularity is achieved.
Pulse Oximetry
Pulse oximetry estimates arterial oxygen saturation by using the characteristic of light absorption on the heme molecule and is the most common technique used to monitor oxygen saturations in the modern NICU. It provides a direct measurement of the percent of oxygen saturation of the red blood cells, but does not take into account anemia, HbF, 2,3-diphosphoglycerate content, oxygen content, and oxygen delivery. (As an example, infants with anemia can have high oxygen saturations even with little total oxygen delivery.) The optimal SpO2 value depends on factors including gestational and postnatal age, clinical diagnosis, temperature, perfusion, movement, and cardiac output. In addition, the performance of SpO2 monitors is not uniform, as described elsewhere.[1,33] Therefore, infants on pulse oximeters must also have their PaO2 levels evaluated and kept within normal ranges.
A valid concern of many neonatal care providers is the risk of possible persistent, significant hypoxemia at low SpO2 ranges in newborns undergoing oxygen therapy. We do not know "exactly" the target saturation range that is optimal for promoting growth and neurologic development while minimizing risk for ROP, BPD, and other adverse oxidative injury. Some clinicians suggest that, based on oxygen content and P50 saturation, a PaO2 of >40 mmHg should be adequate for tissue needs during early neonatal life and during critical illness, given normal hemoglobin concentrations, cardiac output, blood flow, and cellular conditions.[16]
Castillo and associates performed a prospective comparison between SpO2 readings and PaO2 values obtained from arterial samples from newborn infants in seven NICUs. This study confirms that SpO2 levels between 85 and 93 percent correlate with PaO2 levels within the recommended 40--70 mmHg range.[16] With a PaO2 value of 40 mmHg, hemoglobin may be 85 percent saturated. If the hemoglobin concentration is 14 g/dL, the oxygen content would be around 16 mL/dL. This oxygen content is likely to be sufficient to avoid neonatal tissue hypoxia when considering cardiac output, regional blood flow, oxygen consumption, and oxygen delivery.[16]
The American Academy of Pediatrics and others suggest that PaO2 values >80 mmHg may be considered hyperoxemia.[31,34] SpO2 values of >93 percent are frequently associated with PaO2 values of >80 mmHg.[16]
Practice Implications
Oxygen can have potentially significant side effects when inappropiately administered to VLBW infants. Avoiding hypoxia is important, but prolonged hyperoxia and intermittent bouts of hypoxia-hyperoxia can lead to vascular injury and excessive vascular regrowth resulting in ROP.[8] The American Heart Association recently revised its guidelines for neonatal resuscitation to caution the clinician about the excessive use of oxygen in preterm infants. Although supplemental oxygen is recommended whenever positive pressure ventilation is indicated, the revised guidelines recommend that oxygen delivery should be guided by pulse oximetry whenever possible.[35] As Saugstad stated in a review of oxidative stress in the newborn, “We today know that careful control of oxygen levels in the tissues is perhaps the most important means to prevent these conditions, both immediately after birth during resuscitation and in the subsequent neonatal period” (p. 235) (see Table 2 ).[10]
Evidence to support changes in care practices continues to be evaluated. Implementation of better practices has proven to be a challenge in many NICUs. In particular, avoiding periods of hyperoxemia in neonates with constantly fluctuating oxygen saturation levels is both frustrating and time-consuming.
Once a new oxygen saturation policy is in place, what is the compliance of the medical staff in clinical practice? This is the subject of several recent articles.[8,16,36–38] They conclude that success in maintaining intended SpO2 ranges varied substantially among centers, among patients within centers, and for individual patients during their NICU stay. Hagadorn and coworkers showed they were able to keep their sick, oxygen-dependent infants within target range about half of the time. Most of the noncompliance was above the intended range.[38] Clucas and colleagues documented that only 22 percent of their preterm infants' pulse oximetry alarms were set in accordance with protocol.[36]
The challenge of translating the concept of oxygen targeting into practice requires careful planning and education. A better understanding of the relationship of PaO2 and SpO2 as well as the safety and potential benefits of lowered SpO2 targets will increase staff acceptance. As with any practice change, frequent assessment, broad communication, empowerment, and perseverance will be needed to reach the goal of optimum oxygen delivery.
Prognosis
Fortunately, ROP resolves in most cases. "Only 6 percent of infants weighing less than 1,250 g will reach this stage (threshold) of severity, but approximately two-thirds of eyes reaching the 'threshold' will develop severe vision loss or blindness" (p. 913).[23] The current standard of care for the treatment of ROP consists of laser ablation to suppress the neovascular phase of ROP. Laser ablation is expensive and visual outcome is often poor; thus, prevention is still the best strategy. "The World Health Organization's vision 2020 program targets ROP as an "avoidable disease" requiring early detection and treatment to prevent blindness and the inherent costs to the individual and the community. As described by Vision 2020, recent research has resulted in strategies that have been successful in reducing the incidence of ROP as routine fundus examination of premature neonates less than 32 weeks gestation or under 1,250 g and provision of carefully monitored levels of supplemental oxygen" (p. 698).[3]
Summary
In the fragile neonate, the choice of any intervention needs to be carefully considered. A better understanding of the causes of ROP guiding the best care clinical practice in NICUs may greatly decrease the incidence of ROP for neonates in the future. Current research explains how treatment-induced oxygen injury interrupts vascular development and increases the incidence of ROP. Chow and coauthors state, "There is no evidence that VLBW infants need to be managed with an FiO2 that leads to surface oxygen saturation levels of 95 percent to 100 percent. Actually, these levels are potentially dangerous" (p. 344).8 Best care recommendations based on this literature review support a consistent clinical practice of administering the lowest inspired oxygen concentration necessary to prevent hypoxemia. I encourage all neonatal health care providers to examine their practice and to provide precise control of supplemental oxygen to reduce the incidence of retinopathy of prematurity.
References
- Anderson, C., Benitz, W., & Madan, A. (2004). Retinopathy of prematurity and pulse oximetry: A national survey of recent practices. Journal of Perinatology, 24, 164-168.
- Humes, E. (2000). Baby ER (pp. 127-137). New York: Simon and Schuster.
- Wheatley, D. M., Dickenson, J., Mackay, D., Craig, J., & Sale, M. (2002). Retinopathy of prematurity: Recent advances in our understanding. The British Journal of Ophthalmology, 86, 696-701.
- Keshet, E. (2003). Preventing pathological regression of blood vessels. The Journal of Clinical Investigation, 112, 27-29.
- Palmer, E., Flynn, J., Hardy, R., Phelps, D., Phillips, C., Schaffer, D., et al. (1991). Incidence and early course of retinopathy of prematurity. The Cryotherapy for Retinopathy of Prematurity Cooperative Group. Ophthalmology, 98, 1628-1640.
- Tin, W., Milligan, D. W., Pennefather, P., & Hey, E. (2001). Pulse oximetry, severe retinopathy, and outcome at one year in babies of less than 28 weeks gestation. Archives of Disease in Childhood. Fetal and Neonatal Edition, 84, F106-F110.
- York, J. R., Landers, S., Kirby, R., Arbogast, P., & Penn, J. (2004). Arterial oxygen fluctuation and retinopathy of prematurity in very-low-birth-weight infants. Journal of Perinatology, 24, 82-87.
- Chow, L. C., Wright, K. W., Sola, A., & CSMC Oxygen Administration Study Group. (2003). Can changes in clinical practice decrease the incidence of severe retinopathy of prematurity in very low birth weight infants? Pediatrics, 111, 339-345.
- Smith, L. E. (2002). Pathogenesis of retinopathy of prematurity. Acta Paediatrica, 91, 26-28.
- Saugstad, O. (2005). Oxidative stress in the newborn—A 30 year perspective. Biology of the Neonate, 88, 228-236.
- Blackburn, S. T. (2003). Hematologic and hemostatic systems. In Maternal, fetal, & neonatal physiology (2nd ed., pp. 213-254). St. Louis: Saunders.
- Smith, L. E. (2004). Can we restore aspects of the in utero environment in premature infants to prevent disease? Pediatrics, 111 (2 part 1), 491.
- Hellstrom, A., Engstrom, E., Hard, A., Albertsson-Wikland, K., Carlsson, B., Niklasson, A., et al. (2003). Postnatal serum insulin-like growth factor 1 deficiency is associated with retinopathy of prematurity and other complications of premature birth. Pediatrics, 112, 1016-1020.
- Hylander, M., Strabino, D., Pezzullo, J., & Dhanireddy, R. (2001). Association of human milk feedings with a reduction in retinopathy of prematurity among very low birthweight infants. Journal of Perinatology, 21, 349.
- Coalson, J. (2006). Pathology of bronchopulmonary dysplasia. Seminars in Perinatology, 30, 179-183.
- Castillo, A., Sola, A., Baquero, H., Neira, F., Ramiro, A., Deulofeut, R., et al. (2008). Pulse oxygen saturation levels and arterial oxygen tension values in newborns receiving oxygen therapy in the neonatal intensive care unit: Is 85% to 93% an acceptable range? Pediatrics, 121, 882-889.
- Askie, L. M., Henderson-Smart, D., Irwig, L., & Simpson, M. (2003). Oxygen-saturation targets and outcomes in extremely preterm infants. The New England Journal of Medicine, 349, 959-967.
- Chess, P. R., D’Angio, C. T., Pryhuber, G. S., & Maniscalco, W. M. (2006). Pathogenesis of bronchopulmonary dysplasia. Seminars in Perinatology, 30, 171-178.
- McCance, K. L. (1997). Altered cellular and tissue biology. In McCance, K. L., & Huether, S. E. (Eds.), Pathophysiology: The biologic basis for disease in adults and children (3rd ed., pp. 44-81). St. Louis: Mosby.
- Mactier, H., & Weaver, L. T. (2005). Vitamin A and preterm infants: What we know, what we don't know, and what we need to know. Archives of Disease in Childhood. Fetal and Neonatal Edition, 90, F102-F108.
- Johnson, L., Bowen, F. W., & Abbasi, S. (1985). Relationship of prolonged pharmacologic serum levels of vitamin E to incidence of sepsis and necrotizing enterocolitis in infants with birth weight 1500 grams or less. Pediatrics, 75, 619-638.
- Seiberth, V., & Linderkamp, O. (2000). Risk factors in retinopathy of prematurity. A multivariate statistical analysis. Ophthalmologica, 214, 131-135.
- Mills, M. (1999). The eye in childhood. The American Academy of Family Physicians, 60, 907-916.
- Kim, T., Sohn, J., Pi, S., & Yoon, Y. (2004). Postnatal risk factors of retinopathy of prematurity. Pediatric and Perinatal Epidemiology, 18, 130-134.
- Bell, F. E., Strauss, R. G., Widness, J. A., Mahoney, L. T., Mock, D. M., Seward, V. J., et al. (2005). Randomized trial of liberal versus restrictive guidelines for red blood cell transfusion in preterm infants. Pediatrics, 115, 1685-1691.
- Bharwani, S., & Dhanireddy, R. (2008). Systemic fungal infections, a meta-review. Journal of Perinatology, 28, 61-66.
- Erti, T., Gyarmati, J., Gaal, V., & Szabo, I. (2006). Relationship between hyperglycemia and retinopathy of prematurity in very low birth weight infants. Biology of the Neonate, 89, 56-59.
- LIGHT-ROP Cooperative Group. (1999). The design of the multicenter study of light reduction in retinopathy of prematurity (LIGHT-ROP). Journal of Pediatric Ophthalmology and Strabismus, 36, 257-263.
- Primhak, R. (2001). Oxygen saturation and retinopathy of prematurity. [Letters to the editor]. Archives of Disease in Childhood. Fetal and Neonatal Edition, 85, F75.
- Roberton, N. (2001). Two sacred cows of neonatal intensive care. [Letters to the editor]. Archives of Disease in Childhood. Fetal and Neonatal Edition, 85, F75-F76.
- Marlow, N. (2001). High or low oxygen saturation for the preterm baby. Archives of Disease in Childhood. Fetal and Neonatal Edition, 84, F149.
- STOP-ROP Multicenter Study Group. (2000). Supplemental therapeutic oxygen for prethreshold retinopathy of prematurity (STOP-ROP), a randomized, controlled trial. I: Primary outcomes. Pediatrics, 105, 295-310.
- Gerstmann, D., Berg, R., & Haskell, R. (2003). Operational evaluation of pulse oximetry in NICU patients with arterial access. Journal of Perinatology, 23, 378-383.
- American Academy of Pediatrics. (2006). Screening examination of premature infants for retinopathy of prematurity. Pediatrics, 117, 572-576.
- American Academy of Pediatrics and American Heart Association. (2006). Neonatal resuscitation: Textbook (5th ed.). Elk Grove Village, IL: Author.
- Clucas, L., Doyle, L., Dawson, J., Donath, S., & Davis, P. (2006). Compliance with alarm limits for pulse oximetry in very preterm infants. Pediatrics, 119, 1056-1060.
- Coe, K., Butler, M., Reavis, N., Klinepeter, E., Purkey, C., Oliver, T., et al., (2005). Special premie oxygen targeting (SPOT). A program to decrease the incidence of blindness in infants with retinopathy of prematurity. The Journal of Nursing Care Quality, 21, 230-235.
- Hagadorn, J., Furey, A., Nghiem, T., Schmid, C., Phelps, D., Pillers, D., et al. (2006). Achieved versus intended pulse oximeter saturation in infants born less than 28 weeks gestation: the AVIOx study. Pediatrics, 118, 1574-1582.
Authors and Disclosures
Charlene Pollan, RN, MSN, NNP-BC began her nursing career in 1976 in an adult intensive care unit. She then spent several years working in a burn center before she found her passion working with ill and premature infants in the newborn intensive care unit at Utah Valley Regional Medical Center in Provo, Utah. Over the last 19 years, she has worked there as a staff nurse and charge nurse prior to becoming a nurse practitioner. Charlene received her master of science at the University of Utah.
Charlene Pollan, RN, MSN, NNP-BC, E-mail: cpolla@msn.com
Neonatal Network. 2009;28(2):93-101. © 2009 Neonatal Network